![]()
InBios SCoV-2 Ag Detect Rapid Test Instructions

For Use Under Emergency Use Authorization (EUA) OnlyFor in vitro Diagnostic Use OnlyFor Prescription Use Only
INTENDED USE
SCoV-2 Ag Detect™ Rapid Test is a lateral flow immunoassay intended for the qualitative detection of SARS-CoV-2 Nucleoprotein antigen in direct anterior nasal swab specimens from individuals who are suspected of COVID-19 by their healthcare provider within 5 days of symptom onset or from individuals without symptoms or other epidemiological reasons to suspect COVID-19 when tested twice over two or three days with at least 24 hours and no more than 48 hours between tests. Testing is limited to laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, that meet the requirements to perform moderate, high or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.
The SCoV-2 Ag Detect™ Rapid Test does not differentiate between SARS-CoV and SARS CoV-2.
Results are for the identification of the SARS-CoV-2 nucleocapsid protein antigen. The antigen is generally detectable in anterior nasal swab specimens during the acute phase of infection. Positive results indicate the presence of viral antigens, but clinical correlation with past medical history and other diagnostic information is necessary to determine infection status. Positive results do not rule out a bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of disease. Laboratories within the United States and its territories are required to report all results to the appropriate public health authorities.
Negative results should be treated as presumptive and confirmation with a molecular assay, if necessary, for patient management may be performed. Negative results do not rule out SARS-CoV-2 infection and should not be used as the sole basis for treatment or patient management decisions, including infection control decisions. Negative results should be considered in the context of a patient’s recent exposures, history and the presence of clinical signs and symptoms consistent with COVID-19.
For serial testing programs, additional confirmatory testing with a molecular test for negative results may be necessary, if there is a high likelihood of SARS-CoV-2 infection, such as in an individual with a close contact with COVID-19 or with suspected exposure to COVID-19 or in communities with high prevalence of infection. Additional confirmatory testing with a molecular test for positive results may also be necessary, if there is a low likelihood of SARS-CoV-2 infection, such as in individuals without known exposures to SARS-CoV-2 or residing in communities with low prevalence of infection.
Clinical performance may vary depending on the variants circulating at the time of testing. The SCoV-2 Ag Detect™Rapid Test is intended for use by medical professionals or trained operators who are proficient in performing tests and trained clinical laboratory personnel or individuals trained in point of care settings. The SCoV-2 Ag Detect™Rapid Test is only for use under the Food and Drug Administration’s Emergency Use Authorization.
SUMMARY AND EXPLANATION OF THE TEST
SCoV-2 Ag Detect™ Rapid Test is a lateral flow immunoassay intended for the qualitative detection of SARS-CoV-2 Nucleoprotein antigen. The test can be performed using direct nasal samples collected without transport media, requires minimal training to perform, and takes less than 25 minutes to obtain results, making it a suitable diagnostic tool for use in a point-of-care setting.
PRINCIPLE OF THE TEST
SCoV-2 Ag Detect™ Rapid Test is a single-use, qualitative, membrane-based lateral flow immunoassay for detection of SARS-CoV-2 Nucleoprotein antigen. This test may be used with direct nasal swabs respiratory samples collected without transport media.
The rapid test membrane is pre-coated with anti-Nucleoprotein antibodies on the test line region and utilizes a separate control line to assure assay flow and performance. A direct nasal swab specimen is eluted with a proprietary lysis buffer solution directly in the test cassette sample port. Then the eluted sample migrates upward on the membrane to react with the test and control lines.
The viral antigens, if present, bind to the antibody-labeled gold conjugates as the specimen flows upward. Gold conjugates bound to a viral antigen continue to travel upwards and are captured by the test line.
If SARS-CoV-2 Nucleoprotein antigen is present in a patient sample, a red line will appear in the test line region. A red line at the control region should always appear if the assay is performed correctly. The presence of this red control line verifies that proper flow has occurred, and no failure of the gold conjugate has occurred. Refer to the Interpretation of Results section for additional information regarding results analysis.
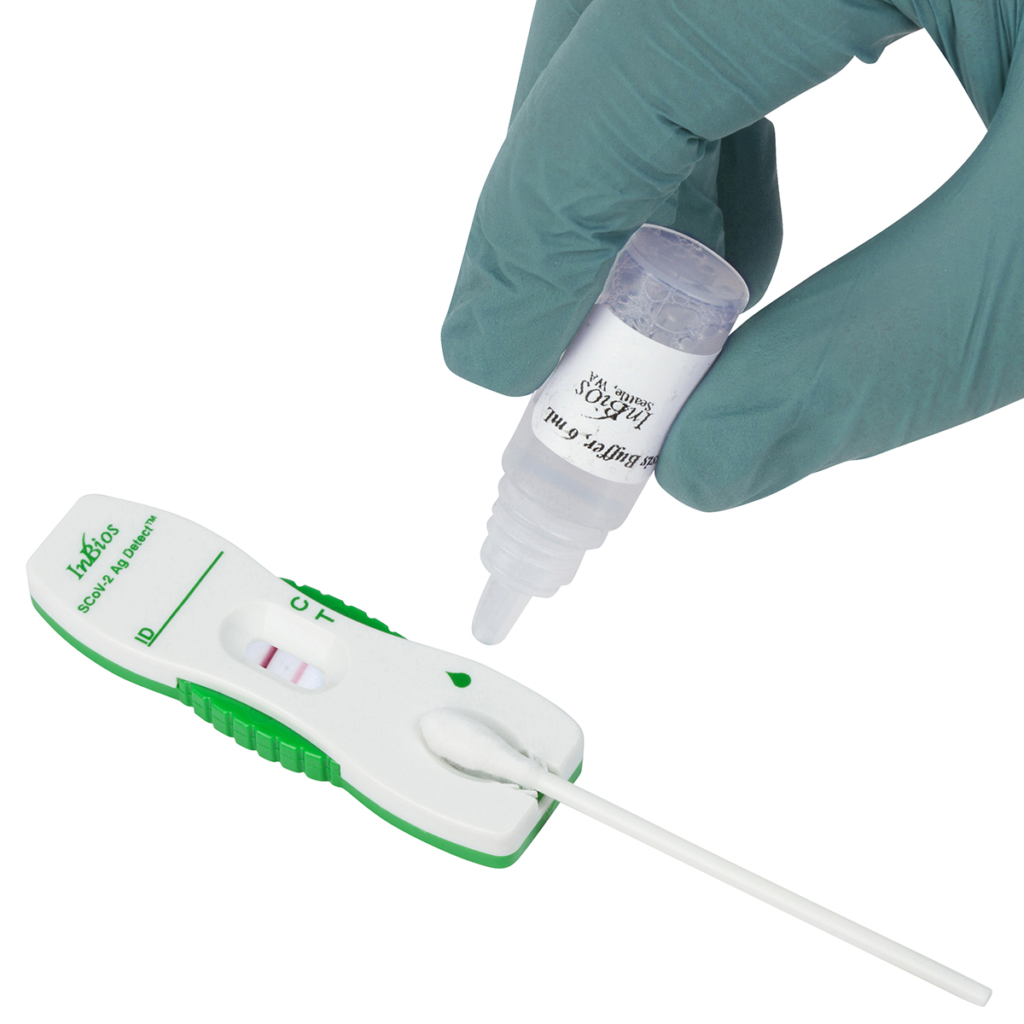
The entire procedure takes approximately 25 minutes. The layout for the SCoV-2 Ag Detect™ Rapid Test is shown below:

PRECAUTIONS
- For prescription and in vitro diagnostic use only.
- In the United States only for use under Emergency Use Authorization. This product has not been FDA cleared or approved, but has been authorized by FDA under an Emergency Use Authorization (EUA) for use by laboratories certified under the CLIA that meet the requirements to perform moderate, high or waived complexity tests. This product is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.
- The emergency use of this product is only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of in vitro diagnostic tests for detection and/or diagnosis of COVID-19 under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization is revoked sooner.
- Laboratories within the United States and its territories are required to report all results to the appropriate public health laboratories.
- This product has been authorized only for the detection of proteins from SARS-CoV-2, not for any other viruses or pathogens.
- Do not use the kit after the expiration date shown on the kit box label.
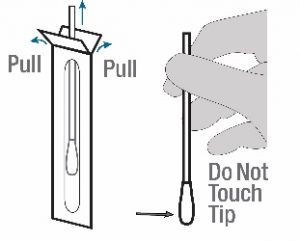
- Leave test cassette sealed in its foil pouch until just before use. The cassette should be used immediately after removal from its pouch to minimize exposure to humidity.Do not use if pouch is damaged or open.
- Proper sample collection, storage and transport are essential for correct results.
- Use only the provided swabs to collect direct nasal swab specimens. Do not use other swabs.
- The provided polyester spun swabs are for single use only. Do not use if there is an evidence of damage or contamination to the swab.
- Do not store or place the swab in the original paper packaging after specimen collection. If storage is needed, use a sterile plastic tube with cap.
- Inadequate or inappropriate sample collection, storage, and transport may yield false test results. Do not store swab specimens in viral transport media or use swab specimens stored in viral transport media with this test.
- Do not use excessive force, pressure or bending when collecting the swab samples as this may result in accidental breakage of the swab shaft.
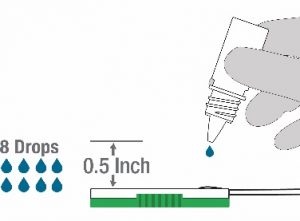
- INVALID RESULTS can occur when an insufficient volume of Lysis buffer is added to the test. To ensure delivery of adequate volume, hold the bottle vertically, ~0.5 inch above the swab well, and add 8 drops slowly. Adding less than 8 drops may result in inaccurate results.
- Wear protective clothing, eye protection, and disposable gloves while performing the assay. Wash hands thoroughly when finished.
- Handle all samples and kits used as if they contain infectious agents. Follow the standard procedures for proper disposal of used kit components.
- Avoid all contact between hands and eyes or mucous membranes during testing.
- Do not eat, drink or smoke in the area where the specimens and kits are handled.
- Do not directly touching the tip of the Lysis buffer bottle to surfaces to avoid contamination. If bottle tip touches any surface, then it is recommended to discard bottle and use a different bottle provided with this kit.
- For best results, perform test interpretation in a well-lit area.
- This is a visually interpreted test. Operators with impaired vision may not be capable of interpreting test results. Individuals with color-impaired vision may not be able to adequately interpret test results.
STORAGE
The kit is designed to be stored at room temperature (15-30°C) for the duration of its shelf life. Exposure to temperatures over 30°C can impact the performance of the test and should be minimized. The kit should not be frozen or refrigerated. The test cassette should be used immediately after removal from its pouch to minimize exposure to humidity.
Human direct nasal swab samples collected without viral transport media can be tested with this assay.
- For information on specimen collection for COVID-19, refer to the following CDC guidelines: Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Persons Under Investigation for Coronavirus Disease 2019 (https://www.cdc.gov/coronavirus/2019-nCoV/lab/guidelines-clinical-specimens.html).
- Specimens should not remain at room temperature (15-30°C) longer than 4 hours, or refrigerated (2-8°C) for longer than 4 days prior to testing.
- If samples are to be shipped, they should be packed in compliance with Federal Regulations covering transportation of infectious agents.
KIT CONTENTS
The kit contains the following components:
- Fifty (50) single-use test cassettes, individually pouched (store at 15-30°C).
- Four (4) dropper bottles of Lysis buffer, 6 mL per bottle (store at 15-30°C).
- Fifty (50) sterile nasal swabs, individually pouched (store at 15-30°C). One of the 50 swabs is intended for use as a negative control.
- Positive Control: swab with SARS-CoV-2 Nucleoprotein antigen dried on the swab. This swab has a blue tint to distinguish it from other swabs in the kit.
- Instructions for use for the SCoV-2 Ag DetectTMRapid Test.
- Quick Reference Instructions for the SCoV-2 Ag Detect™ Rapid Test.
MATERIALS REQUIRED BUT NOT PROVIDED
- Timer
- Pair of gloves
- Biohazard container
TEST PROCEDURE: Sample Collection and Handling
- Collect the direct anterior nasal swab samples following CDC guidelines: Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Persons Under Investigation for Coronavirus Disease 2019 (https://www.cdc.gov/coronavirus/2019-nCoV/lab/guidelines-clinical-specimens.html).
- Wash hands before sample collection. Use appropriate personal protective equipment.

- Remove the swab from the packaging. Be careful not to touch the swab tip (soft end) with hand. If swab tip touches any surface, then it is recommended to discard swab and use a different swab provided with this kit.
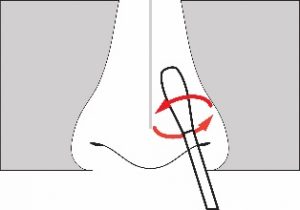
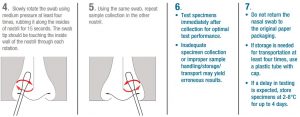
- Carefully insert the swab at least 1 cm (0.5 inch) inside the nostril.
- Slowly rotate the swab using medium pressure at least four times, rubbing it along the insides of nostril for 15 seconds. The swab tip should be touching the inside wall of the nostril through each rotation.
- Using the same swab, repeat sample collection in the other nostril.


- Only the swab provided in the kit is to be used for nasal swab collection.
- Test specimens immediately after collection for optimal test performance. Inadequate specimen collection or improper sample handling/storage/transport may yield erroneous results. Do not return the nasal swab to the original paper packaging. Do not place the swab into transport media.
- If storage is needed for transportation, use a sterile plastic tube with cap without any transport media. Samples are stable for up to 4 hours at room temperature and up to 4 days at 2 -8°C.
TEST PROCEDURE: Specimen Testing

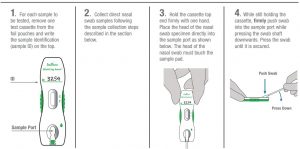
- For each sample to be tested, remove one test cassette from pouch.
- Place the cassette horizontally on a flat surface.
- Write in the specimen ID on the cassette.
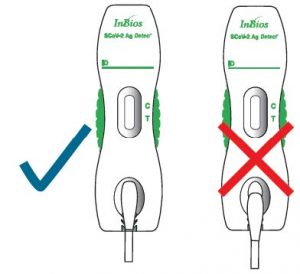
- Use direct anterior nasal swab samples collected following the sample collection steps described in the section above. Test specimens immediately after collection for optimal test performance.Important: Check to be sure the cassette is on a flat surface. False negative results can occur if the test cassette is not on a flat surface.
- Hold the cassette top end firmly with one hand.
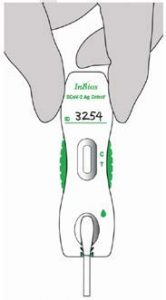
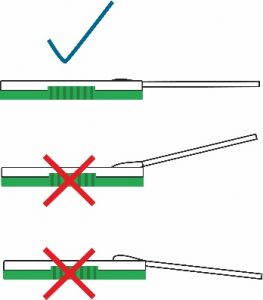
- Place the head of the nasal swab specimen directly into the sample port as shown below. The head of the nasal swab must touch the sample pad at the bottom of the sample port.
- While still holding the cassette, firmly push swab into the sample port while pressing the swab shaft downwards. Press the swab until it is secured.
- The nasal swab should be flat and touchingthe sample pad. Important: Check that the swab covers the sample pad completely. Incomplete coverage of the pad may produce a false negative result.
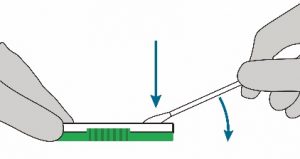
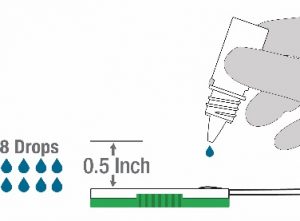
- Hold the Lysis buffer bottle vertically ~0.5inch above the swab head inserted into the sample port.
- SLOWLY add eight (8) drops of Lysis buffer ON TOP of the swab head. ADD ONE (1) drop at a time. DO NOT touch the tip of the dropper bottle to the swab head while dispensing. Important! Adding less than 8 drops may produce invalid results.
- Allow the test cassetteto remain undisturbed. Results should be interpreted between twenty (20) and twenty-five (25) minutes after starting the test. Do not interpret results after twenty-five (25) minutes, as they may be inaccurate.For best results, perform test interpretation in a well-lit area.

 Important: Check that the swab covers the sample pad completely. Incomplete coverage of the pad may produce a false negative result.
Important: Check that the swab covers the sample pad completely. Incomplete coverage of the pad may produce a false negative result.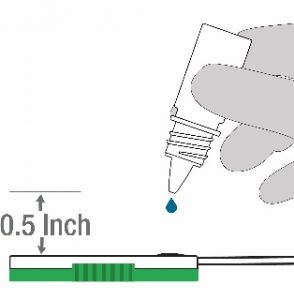
 Important! Adding less than 8 drops may produce invalid results.
Important! Adding less than 8 drops may produce invalid results.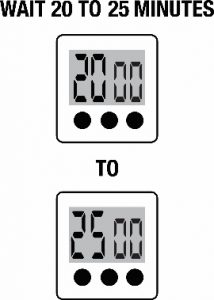
- See the Interpretation of Results section for instructions on how to interpret the SCoV-2 Ag Detect™ Rapid Test results.
- Dispose of all tests and swabs in the appropriate biohazard waste in accordance with federal, state, and local regulations.
TEST PROCEDURE: Positive and Negative Control Testing
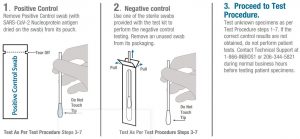
Run the positive and negative controls once per kit upon kit opening to ensure assay integrity. It is recommended that new users run positive and negative controls prior to testing clinical specimens.
- Remove two test cassettes from pouches. Place the cassettes horizontally on a flat surface. Write the control name (i.e. positive control or negative control) on the top of each cassette.
- Place the Positive Control swab (with SARS-CoV-2 Nucleoprotein antigen dried on the swab) into the sample port of one of the cassettes, following steps 5-8 in the section above.
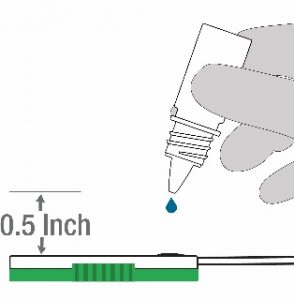
- Hold the Lysis buffer bottle vertically~0.5inch above the swab head inserted into the sample port as in step 9 above.
- SLOWLY add eight drops of Lysis buffer ON TOP of the swab head as in step 10 above. Add ONE DROP at a time. DO NOT touch the tip of the dropper bottle to the swab head while dispensing. Important! Adding less than 8 drops may produce invalid results.
- Repeat steps 2-4 with a negative control swab and the other test cassette. Use an unused, sterile swab to perform the negative control testing.
- Allow the cassettes to remain undisturbed. Results should be interpreted between twenty (20) and twenty-five (25) minutes after starting the test. Do not interpret results after twenty-five (25) minutes, as they may be inaccurate.
- See the Interpretation of Results section for instructions on how to interpret the SCoV-2 Ag Detect™ Rapid Test results.
- If the correct control results are not obtained, do not perform patient tests. Contact Technical Support at 1-866-INBIOS1 or 206-344-5821 during normal business hours before testing patient specimens.A ‘blue’ background color may be present with the Positive Control swab; this is normal and acts as a verification for which swab is the Positive Control.
- Dispose of all tests and swabs in the appropriate biohazard waste in accordance with federal, state, and local regulations.


 Important! Adding less than 8 drops may produce invalid results.
Important! Adding less than 8 drops may produce invalid results.
INTERPRETATION OF RESULTS
The SCoV-2 Ag Detect™ Rapid Test should be interpreted between twenty (20) and twenty-five (25) minutes after starting the test. Do not interpret results after twenty-five (25) minutes because results interpreted after twenty-five (25) minutes may be inaccurate. For best results, perform test interpretation in a well-lit area.
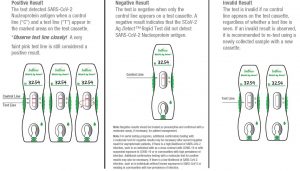
Positive Result: The test detected SARS-CoV-2 Nucleoprotein antigen when a control line (“C”) and a test line (“T”) appear in the marked areas on the test cassette.
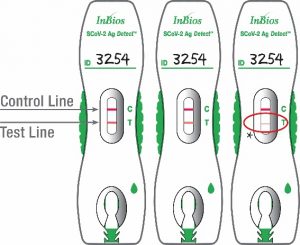
The figures to the right show examples of positive results from specimens, in which SARS-CoV-2 Nucleoprotein antigen was detected.
*Observe Test Line Closely! A very faint pink test line is still considered a positive result.
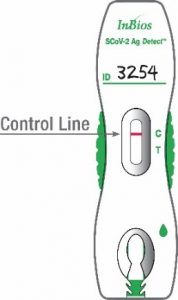
Negative Result: The test is negative when only the control line appears on a test cassette. A negative result indicates that the SCoV-2 Ag Detect™ Rapid Test did not detect SARS-CoV-2 Nucleoprotein antigen. The figure to the right indicates the result from a specimen in which SARS-CoV-2 Nucleoprotein antigen was not detected.
Note: Negative results should be treated as presumptive and confirmed with a molecular assay, if necessary, for patient management.
Note: For serial testing programs, additional confirmatory testing with a molecular test for negative results may be necessary after second negative result for asymptomatic patients, if there is a high likelihood of SARS-CoV-2 infection, such in an individual with as a close contract with COVID-19 or with suspected exposure to COVID-19 or in communities with high prevalence of infection. Additional confirmatory testing with a molecular test for positive results may also be necessary, if there is a low likelihood of SARS-CoV-2 infection, such as in individuals without known exposures to SARS-CoV-2 or residing in communities with low prevalence of infection.
Invalid Result: The test is invalid if no control line appears on the test cassette, regardless of whether a test line is seen. If an invalid result is observed, it is recommended to re-test using a newly collected sample with a new cassette. The figures to the right show invalid results.

LIMITATIONS
- This test detects both viable (live) and non-viable SARS-CoV and SARS-CoV-2. Test performance depends on the amount of virus (antigen) in the sample and may or may not correlate with viral culture results performed on the same sample.
- Failure to follow the instructions for use may adversely affect test performance and/or invalidate the test result.
- False negative test results may occur if a specimen is improperly collected, transported, or handled.
- INVALID RESULTS can occur when an insufficient volume of Lysis buffer is added to the test. To ensure delivery of adequate volume, hold the bottle vertically, ~0.5 inch above the swab well, and add 8 drops slowly. Adding less than 8 drops may result in inaccurate results.
- False negative results can occur when the order of test steps is not correctly followed. Always add the swab to the sample port, and then add lysis buffer to the swab head on test cassette.
- False negative results can occur when the swab is not properly inserted into the test cassette. Be careful to ensure the swab is in full contact with the test cassette prior to proceeding with testing.
- False negative results can occur if the cassette is not placed on a flat surface.
- Performance has only been established with human direct anterior nasal swab specimens without viral transport media using the swab provided. Other specimen types have not been evaluated and should not be used with this assay.
- A negative test result may occur if the level of antigen in a sample is below the detection limit of the test.
- Positive test results do not rule out co-infections with other pathogens.
- Positive test results do not differentiate between SARS-CoV and SARS-CoV-2.
- False negative results are more likely after five days or more of symptoms.
- Negative results are presumptive, do not rule out COVID-19 infection and it may be necessary to obtain additional testing with a molecular assay, if needed for patient management.
- Negative test results are not intended to rule in other non-SARS viral or bacterial infections.
- Freezing and thawing of respiratory specimens must be avoided.
- The performance of this test has not been evaluated for use in patients without signs and symptoms of respiratory infection or other epidemiological reasons to suspect COVID-19, or for serial screening applications tested twice over two or three days with at least 24 hours and no more than 48 hours between tests, and performance may differ in these populations. A study to support use for serial testing will be completed.
- False positive or false negative results may occur.
- If the differentiation of specific SARS viruses and strains is needed, additional testing, in consultation with state or local public health departments, is required.
- The performance of this test was established based on the evaluation of a limited number of clinical specimens collected from February 2021 to April 2021. The clinical performance has not been established in all circulating variants but is anticipated to be reflective of the prevalent variants in circulation at the time and location of the clinical evaluation. Performance at the time of testing may vary depending on the variants circulating, including newly emerging strains of SARS-CoV-2 and their prevalence, which change over time.
The SCoV-2 Ag DetectTM Rapid Test Letter of Authorization, along with the authorized Fact Sheet for Healthcare Providers, the authorized Fact Sheet for Patients, and authorized labeling are available on the FDA website: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-antigen-diagnostic-tests-sars-cov-2
However, to assist clinical laboratories using the SCoV-2 Ag DetectTM Rapid Test (“your product” in the conditions below), the relevant Conditions of Authorization are listed below.
- Authorized laboratories1 using your product must include with test result reports, all authorized Fact Sheets. Under exigent circumstances, other appropriate methods for disseminating these Fact Sheets may be used, which may include mass media.
- Authorized laboratories using your product must use your product as outlined in the authorized labeling. Deviations from the authorized procedures, including the authorized instruments, authorized extraction methods, authorized clinical specimen types, authorized control materials, authorized other ancillary reagents and authorized materials required to use your product are not permitted.
- Authorized laboratories that receive your product must notify the relevant public health authorities of their intent to run your product prior to initiating testing.
- Authorized laboratories using your product must have a process in place for reporting test results to healthcare providers and relevant public health authorities, as appropriate.
- Authorized laboratories must collect information on the performance of your product and report to DMD/OHT7-OIR/OPEQ/CDRH (via email: ) and InBios International Inc. Technical Support (https://inbios.com/technical-support/) any suspected occurrence of false positive or false negative results and significant deviations from the established performance characteristics of your product of which they become aware.
- All operators using your product must be appropriately trained in performing and interpreting the results of your product, use appropriate personal protective equipment when handling this kit, and use your product in accordance with the authorized labeling.
- InBios International Inc. authorized distributors, and authorized laboratories using your product must ensure that any records associated with this EUA are maintained until otherwise notified by FDA. Such records will be made available to FDA for inspection upon request.
1The letter of authorization refers to, “Laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, that meet the requirements to perform moderate, high or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation” as “authorized laboratories.”
PERFORMANCE CHARACTERISTICS
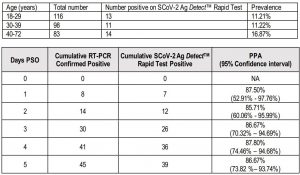
Clinical Performance: Prospective StudyThe clinical performance of SCoV-2 Ag DetectTM Rapid Test was evaluated in a multi-site prospective study in the U.S. In the prospective study, patients presenting with symptoms consistent with possible COVID-19 infection within 5 days of symptom onset were sequentially enrolled. A total of three (3) investigational sites encompassing six (6) drive through collection sites participated in the prospective study.
At the POC sites, paired anterior nasal swabs were collected from eligible patients. One nasal swab was tested with the SCoV-2 Ag DetectTM Rapid Test immediately after collection (i.e. without placing in VTM). The other nasal swab was tested with the comparator assay, an EUA authorized RT-PCR assay. The anterior nasal swabs were assigned to SCoV-2 Ag DetectTM Rapid Test and comparator assay randomly. SCoV-2 Ag DetectTM Rapid Test was performed sequentially by minimally trained operators who are representative of the intended users. Operators only used the Quick Reference Instructions (QRI) for the test without any training provided.
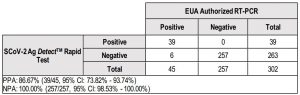
Positive percent agreement (PPA) and negative percent agreement (NPA) from 53 patients enrolled at Site 1, 177 patients enrolled at Site #2 and 73 patients enrolled at Site #3 were evaluated. One specimen was interpreted as invalid on the RT-PCR comparator assay and thus this patient is not included in the analysis.NPA was 100.0% (95% CI: 98.53% – 100.00%) and the PPA was 86.67% (95% CI: 73.82% – 93.74%) for nasal swab samples collected from 302 symptomatic patients within 5 days post symptom onset (PSO).
Patient Demographics
The performance of this test has not yet been clinically validated for use in patients without signs and symptoms of respiratory infection or for serial screening applications and performance may differ in these populations.
Analytical Sensitivity: Limit of Detection (LoD)A limit of detection (LoD) study was conducted to determine the lowest concentration of inactivated SARS-CoV-2 virus in nasal swab matrix at which greater than or equal to 95% of all replicates test positive with the SCoV-2 Ag DetectTM Rapid Test. Limit of detection is 6.3E+03 TCID50/mL.
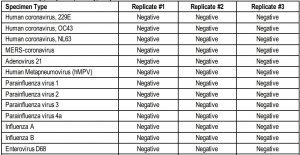
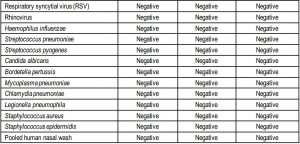
Cross-reactivityThe purpose of this study was to assess whether SCoV-2 Ag Detect™ Rapid Test reacts with related pathogens, high prevalence disease agents, and normal or pathogenic microflora that may be present in clinical nasal swab specimens.
Organisms were evaluated for cross-reactivity by wet testing with the SCoV-2 Ag DetectTM Rapid Test. The potential cross-reactive organisms were spiked into pooled, negative nasal swab matrix at 1E+06 CFU/mL for bacteria/fungi and 1E+05 TCID50/mL or CEID50/mL for viruses. OC43 and parainfluenza virus 4a were tested at lower concentrations (8.9E+04 and 1.6E+04 TCID50/mL, respectively) because the commercially supplied stocks were less than 1E+05 TCID50/mL. The results of this study are shown in the table below.
Cross-reactivity (analytical specificity) study results
The following pathogens were analyzed in silico for sequence homology via NCBI’s BLAST, because they were not available for wet testing.
- Human coronavirus HKU1
- SARS-CoV-1
- Mycobacterium tuberculosis
- Pneumocystis jirovecii (PJP)
The nucleocapsid protein (NP) of human coronavirus HKU1 was determined to have 34% homology with SARS-CoV-2 NP, suggesting a low probability of cross-reactivity. The NP protein of SARS-CoV-1 was determined to have 91% homology with SARS-CoV-2 NP, suggesting cross-reactivity may occur. BLASTs of the Mycobacterium tubercolosis and Pneumocystis jirovecii (PJP) proteomes found no homology, indicating a low probability of cross-reactivity.
The SCoV-2 Ag Detect™ Rapid Test showed no cross-reactivity against samples spiked with other coronaviruses, other respiratory infections which may present with similar symptoms as SARS-CoV-2, or with pooled human nasal wash. SARS-CoV-1 was predicted to be cross-reactive based on protein sequence homology.
Endogenous Interfering SubstancesA study to determine the effects of potentially interfering substances on the SCoV-2 Ag Detect™ Rapid Test was conducted. The interfering substances were mixed either with negative pooled nasal matrix, or with SARS-CoV-2 in pooled negative nasal matrix to yield a final concentration of SARS-CoV-2 of 1.9E+04 TCID50/mL (3x LoD). Each was tested in triplicate. A summary of the results observed is shown below.
Interfering substances study results
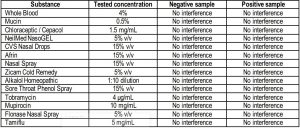
No interference was observed with the SCoV-2 Ag Detect™ Rapid Test for samples that contained blood components and common nasal treatments, or with pooled human nasal wash.
High-dose Hook EffectHook effect was not observed for SCoV-2 in nasal swab matrix in any neat or diluted preparation of SCoV-2 virus for the SCoV-2 Ag DetectTM Rapid Test, up to a concentration of 2.8E+06 TCID50/mL.
![]()
![]()
InBios International, Inc.307 Westlake Ave N, Suite 300Seattle, WA 98109 USA1-866-INBIOS1 (Toll-free USA)+1-206-344-5821 (International)www.inbios.com
Part Number: XXXXXEffective Date: TBD
Quick Reference Instructions
- For Emergency Use Authorization (EUA) Only
- For in vitro Diagnostic (IVD) Use
- For Prescription Use Only
Study the Instructions for Use thoroughly before using Quick Reference Instructions. This is not a complete package insert.
Kit Materials
- Fifty (50) single-use test cassettes, individually pouched (store at 15-30°C).
- Four (4) dropper bottles of Lysis buffer, 6 mL per bottle (store at 15-30°C).
- Fifty (50) sterile nasal swabs, individually pouched (store at 15-30°C). One of the 50 swabs is intended for use as a negative control.
- Positive Control: swab with SARS-CoV-2 Nucleoprotein antigen dried on the swab. This swab has a blue tint to distinguish it from other swabs in the kit.
- Instructions for use for SCoV-2 Ag Detect™ Rapid Test.
- Quick Reference Instructions for SCoV-2 Ag Detect™ Rapid Test.
Test Procedure
- DO NOT open the foil pouch containing the Test Cassettes until ready to test the sample. Place the Test Cassettes on a clean and flat surface, such as a table top.
- Only perform the test within the recommended temperature, 59-86ºF (15-30ºC) and relative humidity, 20% to 85% non-condensing, may cause erroneous results. Repeat the test if it is performed outside these ranges.
- Use direct nasal swab samples collected following the sample collection steps described in the section below.
- Test specimens immediately after collection for optimal test performance.
- Bring samples to room temperature prior to testing.
- For best results, perform test interpretation in a well-lit area.

Interpretation of ResultsDo not interpret results after twenty-five (25) minutes. Results interpreted after 25 minutes may result in false positive, false negative, or invalid result. For best results, perform test interpretation in a well-lit area.
Quality Control
The positive and negative controls should be run:
- Once per kit upon kit opening.
- Once for each new operator.
- As deemed additionally necessary by your internal quality control procedures and in accordance with Local, State and Federal regulations or accreditation requirements.
- Kit includes one set of controls. Additional controls may be purchased separately.
Preparing the positive and negative controls
Sample Collection Procedure
- Collect the direct nasal swab samples following CDC guidelines: Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Persons Under Investigation for Coronavirus Disease 2019 (https://www.cdc.gov/coronavirus/2019-nCoV/lab/guidelines-clinical-specimens.html).
- Use appropriate personal protective equipment.
- Only the swab provided in the kit is to be used for nasal swab collection.

Limitations
Reference the Instructions for Use for Warnings and Precautions, Specimen Collection and Handling, and Quality Control.
- Failure to follow the test procedure may cause erroneous results or invalidate the test results.
- Freezing and thawing of respiratory specimens must be avoided.
- False negative test results may occur if a specimen is improperly collected, transported, or handled.
In the USA, this product has not been FDA cleared or approved; but has been authorized by FDA under an EUA for use by authorized laboratories; use by laboratories certified under the CLIA, 42 U.S.C. §263a, that meet requirements to perform moderate, high or waived complexity tests. This product is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.
This product has been authorized only for the detection of proteins from SARS-CoV-2, not for any other viruses or pathogens; and in the USA, – the emergency use of this product is only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of in vitro diagnostics for detection and/or diagnosis of the virus that causes COVID-19 under Section 564(b)(1) of the Federal Food, Drug and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless declaration is terminated or the authorization is revoked sooner.
Intended Use
SCoV-2 Ag Detect™ Rapid Test is a lateral flow immunoassay intended for the qualitative detection of SARS-CoV-2 Nucleoprotein antigen in direct anterior nasal swab specimens from individuals who are suspected of COVID-19 by their healthcare provider within 5 days of symptom onset or from individuals without symptoms or other epidemiological reasons to suspect COVID-19 when tested twice over two or three days with at least 24 hours and no more than 48 hours between tests. Testing is limited to laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, that meet the requirements to perform moderate, high or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.
The SCoV-2 Ag Detect™ Rapid Test does not differentiate between SARS-CoV and SARS CoV-2.
Results are for the identification of the SARS-CoV-2 nucleocapsid protein antigen. The antigen is generally detectable in anterior nasal swab specimens during the acute phase of infection. Positive results indicate the presence of viral antigens, but clinical correlation with past medical history and other diagnostic information is necessary to determine infection status. Positive results do not rule out a bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of disease. Laboratories within the United States and its territories are required to report all results to the appropriate public health authorities.
Negative results should be treated as presumptive and confirmation with a molecular assay, if necessary, for patient management may be performed. Negative results do not rule out SARS-CoV-2 infection and should not be used as the sole basis for treatment or patient management decisions, including infection control decisions. Negative results should be considered in the context of a patient’s recent exposures, history and the presence of clinical signs and symptoms consistent with COVID-19.
For serial testing programs, additional confirmatory testing with a molecular test for negative results may be necessary, if there is a high likelihood of SARS-CoV-2 infection, such as in an individual with a close contact with COVID-19 or with suspected exposure to COVID-19 or in communities with high prevalence of infection. Additional confirmatory testing with a molecular test for positive results may also be necessary, if there is a low likelihood of SARS-CoV-2 infection, such as in individuals without known exposures to SARS-CoV-2 or residing in communities with low prevalence of infection.
Clinical performance may vary depending on the variants circulating at the time of testing. The SCoV-2 Ag Detect™ Rapid Test is intended for use by medical professionals or trained operators who are proficient in performing tests and trained clinical laboratory personnel or individuals trained in point of care settings. The SCoV-2 Ag Detect™ Rapid Test is only for use under the Food and Drug Administration’s Emergency Use Authorization.
Assistance
If you have any questions regarding the use of this product or if you want to report a problem with the test, please call InBios International, Inc. Technical Support at 1-866-INBIOS1 or 206-344-5821, or visit inbios.com/technical-support/ to submit your inquiry.
![]()
![]() InBios International, Inc.307 Westlake Ave. N., #300Seattle, WA 98109www.inbios.com
InBios International, Inc.307 Westlake Ave. N., #300Seattle, WA 98109www.inbios.com
References
Interim Guidelines for Clinical Specimens for COVID-19 | CDC
HHS Accessibility & Section 508 | HHS.gov
Technical Support – InBios International, Inc.
InBios International, Inc. – Innovative Diagnostics
Technical Support – InBios International, Inc.
InBios International, Inc. – Innovative Diagnostics
[xyz-ips snippet=”download-snippet”]