
![]() GenBody COVID-19 AgRapid Diagnostic Test for the Detection of SARS-CoV-2 AntigenInstructions for Use(IFU)
GenBody COVID-19 AgRapid Diagnostic Test for the Detection of SARS-CoV-2 AntigenInstructions for Use(IFU)
For use under the Emergency Use Authorization OnlyFor in vitro Diagnostic UseFor Prescription Use only
INTENDED USE
The GenBody COVID-19 Ag is an immunochromatographic rapid diagnostic test (RDT) intended for the qualitative detection of nucleocapsid protein antigen from SARS-CoV-2 in direct nasopharyngeal swab (NP) specimens from individuals who are suspected of COVID-19 by their healthcare provider within the first six days of symptom onset.Testing is limited to laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, that meet the requirements to perform moderate, high or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., inpatient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance or Certificate of Accreditation.
Results are for the identification of SARS-CoV-2 nucleocapsid antigen. The antigen is generally detectable in nasopharyngeal swab specimens during the acute phase of infection. Positive results indicate the presence of viral antigens, but clinical correlation with patient history and other diagnostic information is necessary to determine infection status. Positive results do not rule out bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of the disease. Laboratories within the United States and its territories are required to report all positive results to the appropriate public health authorities. Laboratories within the United States and its territories are required to report all results to the appropriate public health authorities.
Negative results should be treated as presumptive and may be confirmed with a molecular assay, if n e necessary, for patient management. Negative results do not rule out SARS-CoV-2 infection and should not be used as the sole basis for treatment or patient management decisions, including infection control decisions. Negative results should be considered in the context of a patient’s recent exposures, history, and the presence of clinical signs and symptoms consistent with COVID-19.
The GenBody COVID-19 Ag is intended for use by medical professionals or operators trained in performing tests in point of care settings. The GenBody COVID-19 Ag is only for use under the Food and Drug Administration’s Emergency Use Authorization.
EXPLANATION OF THE TEST
COVID-19 (short for ‘Coronavirus Disease 2019’) is a disease first recognized in 2019 that is caused by a type of novel coronavirus called SARS-CoV-2. Due to its rapid spread, the World Health Organization (WHO) recognized the disease as a global pandemic on March 11, 2020. Individuals infected with SARS-CoV-2 may have a range of symptoms from asymptomatic infection to severe respiratory illness and even death. The virus is spread primarily from person to person through respiratory particles, even by individuals without symptoms.
The GenBody COVID-19 Ag Test is a rapid, qualitative immuno-chromatographic assay for the determination of the presence of SARS-CoV-2 antigens in human nasopharyngeal swab specimens. The test strip in each device contains mouse monoclonal antibodies to the nucleoprotein (NP) of SARS-CoV-2. When the sample contains SARS-CoV-2 antigens, anti-SARS-CoV-2 monoclonal antibodies that are coupled with colloidal gold bind to SARS-CoV-2 antigens in the sample to form an antigen-antibodycomplex. This complex is then captured by anti-SARSCoV-2 monoclonal antibodies immobilized on the Test line, and a visible line appears on the membrane, while unbounddye complete yes continues to migrate beyond the test line area. Unbound protein-dye complexes are later captured at the Control line. The formation of the Control line serves as an internal control. If the control line does not appear within the designated incubation time (i.e., 15 – 20 minutes), the result is invalid and the test should be repeated with a new sample.
MATERIALS PROVIDED
| Kit Component | Quantity | Description |
| GenBodyCOVID-19 Ag Test Device | Twenty-five (25) single-use Test Devices | individually pouched devices with a desiccant. Test Device contains one reactive test strip.coated with mouse anti-SARS-CoV-2 NP and mouse anti-Nus tag antibodies for the controlimpregnated with Mouse anti-SARS-CoV-2 NP Nus tag antigens |
| The test strip contains membrane antibodies for the test line, and conjugate pad antibodies, and recombinant | ||
| Extraction Solution | Two (2) bottles containing9 mL of Extraction Solution | Buffer with detergent and preservative (< 0.1% sodium azide) |
| Extraction Tube | Twenty-five (25) single-use tubes | Flexible plastic tube for extraction of sample |
| Dropper Tips | Twenty-five (25) single-use dropper tips | Disposable of the Extraction Tube for dispensing the extracted sample |
| Sterilized Nasopharyngeal Swabs | Twenty-five (25) single-use specimen sampling swabs | Swab for nasopharyngeal sample collection with a flexible/breakable handle |
| External Positive Control Swab | One (1) single-use swab | Individually pouched swab coated with non-infectious recombinant SARS-CoV-2 proteinan antigen on the head |
| External Negative Control Swab | One (1) single-use swab | Individually pouched swab coated with the buffer on the head |
| instructions for Use (IFU) | One (1) | Instructions for use |
| Quick Reference Instructions (QRI) | One (1) | Quick reference instructions |
MATERIALS REQUIRED BUT NOT PROVIDED
- Timer
- Any necessary personal protective equipment including gloves
QUALITY CONTROL
Internal Quality ControlEach GenBody COVID-19 Ag Test Device has a built-in internal procedural control. The reddish-purple line appearing at the “C” position is internal procedural control. This procedural control line indicates that sufficient flow has occurred, and the functional integrity of the test device has been maintained. A distinct reddish-purple Control line should always appear if the test has been performed correctly. If the control line does not appear, the test result is invalid and a new test should be performed.
External Quality ControlGood laboratory practice includes the use of external controls to ensure proper kit performance. It is recommended that external control testing be performed with each new operator and before using a new lot or shipment of GenBody COVID-19 Ag kits to confirm the expected QC results, using the external controls provided in the kit. The frequency of additional QC tests should be determined according to your laboratory’s standard QC procedures and local, State, and Federal regulations or accreditation requirements. Upon confirmation of the expected results, the kit is ready for use with patient specimens. The GenBody COVID-19 Ag kit contains two control swabs. Test the control swabs in the same manner as patient specimens. When the positive control is tested, reddish-purple lines appear at the C and T positions. When the negative control is tested, a reddish-purple line appears at the C position only. If external controls do not perform as expected, do not use the test results and contact Technical Support at (888) 552-5204 or [email protected].The use of positive and negative controls from other commercial kits has not been established with the GenBody COVID-19 Ag test.
SPECIMEN COLLECTION AND STORAGE
Swab Sample Collection ProcedureOnly the swab provided in the kit is to be used for swab sample collection. If a deviated septum or blockage creates difficulty in obtaining the specimen from one nostril, use the same swab to obtain the specimen from the other nostril. Refer to the CDC Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Persons for Coronavirus Disease 2019 (COVID-19) (https://www.cdc.gov/coronavirus/2019-ncov/lab/guidelinesclinical-specimens.html).
Nasopharyngeal Swab Sample Collection Procedure 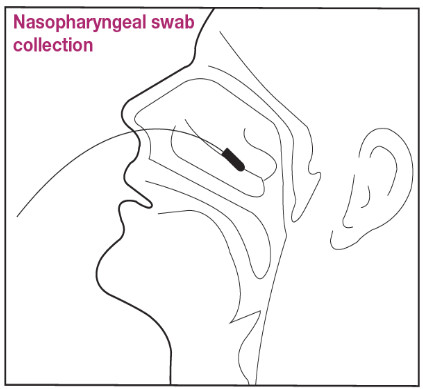
- Remove a nasopharyngeal swab from the pouch.
- With the patient’s head tilted backward at 70 degrees, carefully insert the swab into the nostril that presents the most secretion under visual inspection.
- Gently and slowly insert the swab through the nostril parallel to the palate (not upwards) until resistance is encountered or the distance is equivalent to that from the ear to the nostril of the patient, indicating contact with the nasopharynx.
- Leave the swab in place for several seconds to absorb secretions.
- Rotate the swab 3-5 times against the posterior nasopharynx.
- Using gentle rotation, remove the swab from the nostril; insert into the Extraction Tube.
- All specimens should be tested as soon as they are prepared.
Specimen Storage and Handling ProcedureFor the best performance, swab specimens collected from patients should be tested immediately after collection. The collected swab specimen can be tested for up to 60 minutes, following sample collection. If the sample is extracted from the swab, the extracted sample can be tested for up to 5 hours if stored between 2-30 °C.
TEST PROCEDURES
Procedural Notes• Allow Test Devices, reagents, specimens, and/or controls to equilibrate to room temperature (15~30°C) prior to testing.• Do not open the foil pouch until one is ready to perform the test.• Several tests may be run at one time.• Label the device with the patient identification or control to be tested.• Place Test Device on a level surface.• Used samples, swabs, tubes, and Test devices should be treated as biohazardous waste.
Specimen Swab Test Procedure
| Step 1 | 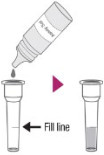 |
Add the Extraction Solution to the Fill Line indicated on the Extraction Tube (400 µL). |
| Step 2 |  |
A. Insert the collected specimen swab into theExtraction Solution.B. Mix by squeezing the tube and simultaneously rotating the Swab 8 – 10times. Remove the swab from the Extraction Tube while pressing the swab against the side of the tube to extract the solution. |
| Step 3 | 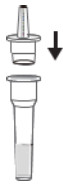 |
Place the Dropper Tip on the Extraction Tube. |
| Step 4 | 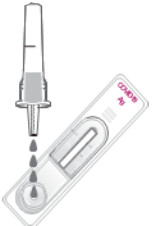 |
Add 4 drops (~100 µL) of the solution to the center of the sample well of the Test Device. |
| Step 5 |  |
Read the test result at 15-20 minutes.Test results should not be read after 20 minutes |
INTERPRETATION OF THE RESULTS

- Positive result: Two reddish-purple lines appear in the test window, one on the test line position (T) and the other on the control line position (C).Note: The test line (reddish-purple line) may vary in shade and intensity (light or dark, weak or strong) depending on the concentration of antigen detected. The intensity of the Control line should not be compared to that of the Test line for the interpretation of the test result. Any faint visible reddish-purple Test line should be interpreted as positive.
- Negative result: Only one reddish-purple line on the control line (C) position appears with no line on the test line position (T).
- Invalid result: If a line does not appear on the control line position (C) in 15 minutes, the test result is invalid. Re-test with a new GenBody COVID-19 Ag Test Device.
STORAGE AND STABILITY
- GenBody COVID-19 Ag kit should be stored between 2 to 30 °C (35.6 to 86 °F).
- Kit components in the Ge body COVID-19 Ag kit are stable until the expiration date printed on the label.
- The Test Device must remain in the sealed foil pouch until use.
WARNINGS & PRECAUTIONS
- For in vitro diagnostic use only.
- This product has not been FDA cleared or approved; but has been authorized by FDA under a EUA for use by laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, to perform moderate, high, or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., inpatient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.
- Federal Law restricts this device to sale by or on the order of a licensed practitioner (the US only).
- This product has been authorized only for the detection of proteins from SARS- CoV-2, not for any other viruses or pathogens.
- The emergency use of this product is only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of in vitro diagnostics for the detection and/or diagnosis of COVID-19 under Section 564(b)(1) of the Federal Food, Drug and Cosmetic Act, 21 U.S.C. § 360bbb-3 (b)(1), unless the declaration is terminated or authorization is revoked sooner.
- Laboratories within the United States and its territories are required to report all results to the appropriate public health laboratories.
- Do not use the kit past its expiration date.
- Do not store or test specimens in viral transport media, as it may result in false-positive or false-negative results.
- Wear appropriate personal protective equipment and gloves when running each test and handling patient specimens. Change gloves between the handling of specimens.
- The Extraction Solution in this kit contains a detergent and a preservative that will inactivate cells and virus particles. Samples eluted in this solution are not suitable for culture.
- Test Devices are single-use only and should be discarded after use. Do not re-use the Test Device.
- Proper sample collection, storage, and transport are essential for correct results. Specimens should be prepared in accordance with the instructions provided in the “Specimen Collection and Storage” section.
- Excess blood or mucus on the swab specimen may interfere with test performance, potentially yielding an inaccurate result. Avoid touching any bleeding areas of the nasopharynx when collecting specimens.
- Users should test specimens as quickly as possible after specimen collection.
- Exposure to humidity may decrease the stability of the reagents. The test should be performed immediately after removing the device from the foil pouch. Do not use if the pouch is damaged or opened.
- Test devices and swabs should be used immediately upon opening; do not remove Test Devices f r om the pouch until just before use.
- Do not use the Test Device if the desiccant included in the foil pouch has changed from yellow to green.
- To ensure delivery of adequate volume, hold the tube vertically and add drops slowly.
- Dispose of used contents as biohazardous wastes in accordance with federal, state, and local requirements.
- Test results must be evaluated in conjunction with other clinical data available to the licensed practitioner.
- Do not use the kit components from different lots.
- Swabs included in the kit are approved for the GenBody COVID-19 Ag test. Do not use other swabs.
- Do not smoke, eat, or drink in areas in which specimens or kit reagents are handled.
- If the extraction solution contacts the skin or eye, flush with copious amounts of water.
- For additional information on safety, handling, and disposal of the components within this kit, including the Safety Data Sheet (SDS), please email or call Technical Support at [email protected] or (888)552-5204.
LIMITATIONS
- This device is for professional in vitro diagnostic use only.
- This device is only used for testing direct human nasopharyngeal swab specimens. Viral transport media (VTM) should not be used with this test.
- This test is not for use in at-home testing settings.
- The amount of antigen in a sample may decrease as the duration of illness increases. Specimens collected after six days are more likely to be negative compared to RT-PCR.
- A negative test result may occur if the level of antigen in a sample is below the detection limit of the test or if the sample was collected improperly.
- The performance of the GenBody COVID-19 Ag was evaluated using the procedures provided in this Instructions for Use (IFU) only. Modifications to these procedures may alter the performance of the test.
- This test detects both viable (live) and non-viable SARS-CoV-2. Test performance depends on the amount of viral antigen in the sample and may or may not correlate with viral culture results performed on the same sample.
- Failure to follow the Test Procedure may adversely affect test performance and/or invalidate the test result.
- Positive test results do not rule out co-infections with other pathogens.
- Negative results do not rule out SARS-CoV-2 infection, particularly in those who have been in contact with the virus. Follow-up testing with a molecular diagnostic should be considered to rule out infection in these individuals.
- Negative test results are not intended to rule in other non-SARS viral or bacterial infections.
- Negative results should be treated as presumptive and confirmed with a molecular assay for clinical management, if necessary.
- The performance of this test was established based on the evaluation of a limited number of clinical specimens collected between January 2021 to February 2021. The clinical performance has not been established in all circulating variants but is anticipated to be reflective of the prevalent variants in circulation at the time and location of the clinical evaluation. Performance at the time of testing may vary depending on the variants circulating, including newly emerging strains of SARS-CoV-2 and theirprevalence, which changes over time.
The GenBody COVID-19 Ag Letter of Authorization, along with the authorized Fact Sheet for Healthcare Providers, the authorized Fact Sheet for patients, and authorized labeling are available on the FDA website:(https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizationsmedical-devices/in-vitro-diagnostics-euas-antigen-diagnostic-tests-sars-cov-2)However, to assist clinical laboratories using the GenBody COVID-19 Ag (“your product” in the conditions below), the relevant Conditions of Authorization are listed below:
A. Authorized laboratories* using your product must include, with test result reports, all Fact Sheets. Under exigent circumstances, other appropriate methods for disseminating these Fact Sheets may be used, which may include mass media.B. Authorized laboratories using your product must use your product as outlined in the authorized labeling. Deviations from the authorized procedures, including the authorized instruments, authorized clinical specimen types, authorized control materials, authorized other ancillary reagents and authorized materials required to use your product are not permitted.C. Authorized laboratories that receive your product must notify the relevant public health authorities of their intent to run your product prior to initiating tests.D. Authorized laboratories using your product must have a process in place for reporting test results to healthcare providers and relevant public health authorities, as appropriate.E. Authorized laboratories must collect information on the performance of your product and report to DMD/OHT7-OIR/OPEQ/CDRH (via email: CDRH-EUA- ) and GenBody Inc. (viaemail: [email protected]) any suspected occurrence of false positive or false negative results and significant deviations from the established performance characteristics of your product of which they become aware.F. All operators using your product must be appropriately trained in performing and interpreting the results of your product. Use appropriate personal protective equipment when handling this kit, and use your product in accordance with the labeling.G. GenBody Inc. and authorized laboratories using your product must ensure that any records associated with this EUA are maintained until otherwise notified by the FDA. Such records will be made available to the FDA for inspection upon request.
*The letter of authorization refers to, “Laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, meet requirements to perform high, moderate, or waived complexity tests. This test is authorized for use at the Point of Care (POC) i.e., inpatient care settings operating under CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation” as “authorized laboratories.”
PERFORMANCE CHARACTERISTICS
a. Analytical Sensitivity: Limit of Detection (LoD)The Limit of Detection (LoD) of the GenBody COVID-19 Ag test was determined using serial dilutions of the heat-inactivated SARS-CoV-2 (USA-WA1/2020). Testing sample was prepared by spiking the strain into the pooled human nasopharyngeal swab matrix obtained from healthy volunteers confirmed negative by RT-PCR. The initially determined LoD by two-fold serial dilution was confirmed by testing in 20 replicates. The confirmed LoD for the GenBody COVID-19 Ag was 1.11 x 10 2 TCID50/mL.b. Analytical Reactivity to VariantsThe analytical reactivity (sensitivity) to the variants of SARS-CoV-2 was tested. The GenBody COVID-1 9 A g test detects the B.1.1.7/UK variant at 6.25 x 10 1 pfu/mL and .1.351/South Africa variant at 4.38 x 10 1 pfu/mL.c. High-dose hook effectThe GenBody COVID-19 Ag was tested up to 1.15 x 107TCID50/mL of heat-inactivated SARS-CoV-2 (USA- WA1/2020) and no high-dose hook effect was observed.d. Analytical Specificity: Cross-reactivity and Microbial interferenceCross-reactivity and interference studies were performed for related pathogens, high prevalence disease agents, and normal or pathogenic flora that are reasonably likely to be encountered in the clinical specimen of the nasal cavity. Each organism and virus (15 bacteria and 29 viruses) was tested in both the absence and presence of inactivated SARS-CoV-2 (SARS-CoV-2 isolate USA-WA1/2020) at the 2x LoD. All testing samples were prepared in the negative clinical nasopharyngeal matrix. No cross-reactivity or interference was observed at the concentration tested shown in the below table.
| Microorganism | Concentration |
| Adenovirus (C1 Ad. 71) | 1.41 x 106TC1D50/mL |
| Enterovirus 068 | 5.01 x 105 TCIDso/mL |
| Human Metapneumovirus (hMPV) | 3.80 x 106TC1D50/ml |
| Influenza A H1N1(New Ca1/20/99) | 1.15 x 107TC1D50/mL |
| Influenza B (Florida/02/06) | 1.41 x 106TC1D50/mL |
| Parainfluenza virus 1 | 9.12 x 108 TCID50/mL |
| Parainfluenza virus 2 | 4.17 x 105TC1D50/mL |
| Parainfluenza virus 3 | 6.61 x 106TC1D50/mL |
| Parainfluenza virus 4A | 1 x 10658 TC1050/mL |
| MERS-coronavirus | 3.55 x 105 TC1D50/mL |
| Human coronavirus 229E | 4.17 x 105TC1D50/mL |
| Human coronavirus0C43 | 1.26 x 106TC1D50/mL |
| Human coronavirus NL63 | 1.41 x 106TC1D50/mL |
| SARS-coronavirus (in PBS) | 1 x 108 pfu /mL |
| SARS-coronavirus (Vero E6 Cell DMEM) | 1 x 108 pfu /mL |
| Respiratory syncytial virus – Type A | 3.80 x 106TC1D50/mL |
| Respiratory syncytial virus – Type B | 1 x 10 TCIDso/mL |
| RhinovirusType 1A | 1x 10658 TC1050/mL |
| RhinovirusType 14 | 9.8 x 107 pfu/ mL |
| RhinovirusType 42 | 4.2 x 105 pfu / mL |
| Cytomegalovirus | 1 x 107U/ mL |
| Epstein-Barr Virus | 2.70 x 108 cp/ mL |
| Varicella Zoster Virus | 4 x 108cp/ mL |
| Parvovirus B19 | 8 x 108IU/ mL |
| Human Immunodeficiency Virus —1 | 4 x 109IU/ ml |
| Human Immunodeficiency Virus — 2 | 5.6 x 107U/ ml |
| Microorganism | Concentration | |
| Hepatitis B Virus(Performancepanel, Se racare,0805-0362,Batch#10387873) | DNA genotype-A | 5.5 x 1071U/ mL |
| DNA genotype-B | 4.2 x 1051U/ mL | |
| DNA genotype-C | 1.0 x 108 IU/ mL | |
| DNA genotype-D | 3.2 x 1031U/ mL | |
| DNA genotype-E | 3.5 x 1031U/ mL | |
| DNA genotype-F | 1.5 x 105 IU/ mL | |
| DNA genotype-H | 3.0 x 102 IU/ mL | |
| Herpes Simplex Virus-1 | 1 x 106 TCID50/ mL | |
| Herpes Simplex Virus-2 | 1 x 106 U/ mL | |
| Hepatitis C Virus | 1 x 106TCID50/ mL | |
| Candida albicans | 6.27 x 108 CFU/mL | |
| Chlamydia pneumoniae | 2.12 x 1081FU/mL | |
| Haemophilus influenzae | 5.43 x 108 CFU/mL | |
| Legionella pneumophila | 1.63 x 1010 CFU/mL | |
| Mycobacterium tuberculosis | 6.86 x 107 CFU/mL | |
| Mycoplasma pneumoniae | 3.16 x 108 CCU/mL | |
| Pseudomonas aeruginosa | 3.44 x 109 CFU/mL | |
| Staphylococcus epidermidis | 9.27 x 109 CFU/mL | |
| Staphylococcus aureus | 8.5 x 106 CFU/ mL | |
| Streptococcus pneumoniae | 4.16 x 108 CFU/mL | |
| Streptococcus pyogenes | 1.64 x 109 CFU/mL |
| EUA only Streptococcus sali varius | 8.17 x 108 CFU/mL |
| Escherichia coli | 81.3 x 10 CFU/ mL |
| Bordetella pertussis | 1.13 x 1010 CFU/mL |
| Pooled human nasal wash –representative of normal respiratory microbial flora | 100% |
To estimate the likelihood of cross-reactivity with SARS-CoV-2 of organisms that were not available for wet testing, in-silico analysis using the Basic Local Alignment Search Tool (BLAST) managed by the National Center for Biotechnology Information (NCBI) was used to assess the degree of protein sequence homology. HKU1 nucleocapsid phosphoproteins, Mycobacterium tuberculosis, and Pneumocystis jirovecii (PJP) were analyzed and the results are below.
- The homology between SARS-CoV-2 nucleocapsid protein and human coronavirus HKU1 nucleocapsid phosphoproteins is relatively low, at 36.7% across 82% of sequences, but cross-reactivity cannot be ruled out.
- The homology between SARS-COV-2 nucleocapsid protein and Pneumocystis jirovecii (PJP) total protein is relatively low, at 22.0% across 4% of sequences, but cross-reactivity cannot be ruled out.
- No homologous protein sequence was found as a result of in-silico analysis with Mycobacterium tuberculosis total protein and SARS-CoV-2 nucleocapsid protein. Despite there being little homology observed, the c cross-reactivity of GenBody COVID-19 Ag against Mycobacterium tuberculosis cannot be ruled out.
e.Endogenous Interfering SubstancesThe interference study was performed for the 22 potentially interfering substances that may be found in the upper respiratory tract. The positive (2x LoD SARS-CoV-2) and negative samples were tested with the addition of potentially interfering substances. The performance of GenBody COVID-19 Ag was not affected by any of the potentially interfering substances listed in the table below at the concentrations tested.
| Substance | Concentration |
| Whole blood | 5% |
| Nagel (NeilMed) | 5% v/v |
| Phenylephrine (Nasal Drop) | 10% v/v |
| Acetylsalicylic acid | 20 mg/ml |
| Beclomethasone | 0.5 mg/ml |
| Benzocaine (Vicks) | 5% |
| Flunisolide | 3 mg/ml |
| Mucin(Bovine submaxillary gland) | 0.5% |
| Menthol | 10 mg/ ml |
| Oxymetazoline (Afrin) | 15% v/v |
| Tobramycin | 40 mg/ml |
| Substance | Concentration |
| Zanamivir | 3.3 mg/ml |
| Oseltamivir phosphate (Tamiflu) | 12 mg/ml |
| Cromolyn (Nasal Spray) | 40 mg/ ml |
| Homeopathic (Alkalol) | 5% v/v |
| Zicam Cold Remedy | 5% v/v |
| Mucous | 35% v/v |
| Guaiacol glyceryl ether | 20 mg/ml |
| Chloraseptic (Menthol/Benzocaine) | 1.5 mg/mL |
| Chloraseptic spray (phenol) | 15% v/v |
| Mupirocin | 10 mg/mL |
| Fluticasone Propionate | 5% v/v |
CLINICAL EVALUATION
A prospective clinical study was conducted from January 2021 to February 2021 at point of care (POC) sites in the United States to evaluate the performance of the GenBody COVID-19 Ag test for direct nasopharyngeal swab specimens compared to an Emergency Use Authorized (EUA) RT-PCR test. A total of seven (7) operators from three (3) POC sites were involved in the study. Patients were prospectively and sequentially enrolled at each site. Samples were collected from patients of all ages who visited the doctor with signs and symptoms of suspected COVID-19 infection. The performance of the GenBody COVID-19 Ag test was established with 107 nasopharyngeal swab samples collected from patients within 6 days of onset of COVID-19.Two nasopharyngeal swabs were collected from each patient. One nasopharyngeal swab was tested directly using the GenBody COVID-19 Ag test according to the product instructions. The other swab was tested on the comparator RT-PCR. Swabs were randomly assigned to test with the GenBody COVID-19 Ag test or the RT-PCR.
Clinical Study ResultsA. Patient DemographicsThe patient demographic information (age, gender, and elapsed time from date of on-set) is below.
| Age Group | Male | Female | Total | |||
| No. ofsamples | % | No. ofsamples | % | No. ofsamples | % | |
| 5 years of age | 0 | 0.00% | 0 | 0.00% | 0 | 0.00% |
| 6-21 years of age | 10 | 9.% | 12 | 11.% | 22 | 21.% |
| 22-59 years of age | 33 | 31.% | 39 | 36.% | 72 | 67.% |
| a 60 years of age | 5 | 5.% | 8 | 7.% | 13 | 12.% |
| Total | 48 | 45.% | 59 | 55.% | 107 | 100.% |
Table a-1. The specimen positivity breakdown based on age and gender of the patient
| Age Group | GenBody COVID-19 Ag | ||
| Total # | Positive | Prevalence | |
| 55 years of age | 0 | 0 | 0.00% |
| 6-21 years of age | 22 | 9 | 41.% |
| 22-59 years of age | 72 | 22 | 31.% |
| 60 years of age | 13 | 10 | 77.% |
| Total | 107 | 41 | 38.% |
Table a-2. Positive results are broken down by days since symptom onset
| Days SinceSymptom Onset | CumulativeRT-PCRPositive (+) | CumulativeGenBody COVID-19 Ag Positive (+) | PPA | 95% Confidence Interval | |
| 0 | 3 | 2 | 67.% | 9.% | 99.% |
| 1 | 11 | 9 | 82.% | 48.% | 98.% |
| 2 | 19 | 16 | 84.% | 60.% | 97.% |
| 3 | 24 | 21 | 88.% | 68.% | 97.% |
| 4 | 31 | 28 | 90.% | 74.% | 98.% |
| 5 | 42 | 38 | 90.% | 77.% | 97.% |
| 6 | 45 | 41 | 91.% | 79.% | 98.% |
| Total | 45 | 41 | 91.% | 79.% | 98.% |
B. Clinical PerformanceThe performance of the GenBody COVID-19 Ag test at POC sites for samples within 6 days of symptom onset was compared to a EUA RT-PCR test and is presented below. A total of 107 patients was enrolled in the clinical study presented with symptoms within 6 days of onset. 41/45 (PPA 91.1%) samples tested positive and 62/62 (NPA 100%) samples tested negative when compared with the comparator.Table b-1 presents the summary of the GenBody COVID-19 Ag test performance data compared to the comparator EUA RT-PCR test when all data from 3 POC sites are combined.Table b-1. Summary of the performance of GenBody COVID-19 A g compared to RT-PCR for all sites
| All Sites | RT- PCR | |||
| Positive | Negative | Total | ||
| GennadyCOVID-19 Ag | Positive | 41 | 0 | 41 |
| Negative | 4 | 62 | 66 | |
| Total | 45 | 62 | 107 |
| Estimate | 95% CI | ||
| LCI | UCI | ||
| Sensitivity (% PPA) | 91.% | 79.% | 98.% |
| Specificity (%NPA) | 100% | 94.% | 100% |
| Positive Predictive Value (PPV) | 100% | 91.% | 100% |
| Negative Predictive Value (NPV) | 94.% | 85.% | 98.% |
| Prevalence | 42.% | 33.% | 52.% |
PERFORMANCE WITH ANALYTE CONCENTRATION NEAR THE LoD CONCENTRATION
To demonstrate that non-laboratory personnel can perform the GenBody COVID-19 Ag test accurately with weak positive samples in the intended use environment, a study was performed at 3 point of care (POC) sites by the sting positive samples at 2x LoD and negative samples. A total of 6 operators who were medical assistants or nursesparticipated in the study (2 operators at each site).Each operator performed tests blindly using the coded samples. All operators performed the GenBody COVID-19 Ag test accurately (100 % agreement with expected results) in the intended use environment.
TECHNICAL SUPPORT
For questions, or to report a problem, please call Technical Support at (888) 552-5204 (Available Hours: Mon. to Fri.: 9 a.m. – 5 p.m. PST) or [email protected].Test system problems may also be reported to the FDA using the MedWatch reporting system (phone: 1-800 FDA-1088; fax: 1-800 FDA-1078: or http://www.fda.gov/medwatch).
ORDERING AND CONTACT INFORMATION
Kwell Laboratories, LLC (US Distributor/US Agent)Tel: (949) 561-0664Email: [email protected]
INTERNATIONAL SYMBOL USAGE
You may see one or more of these symbols on the labeling/packaging of this product:
 |
Use-by date | Batch Code | In vitro diagnostic device | ||
| Catalog number |  |
Consult instructions for use |  |
Manufacturer | |
| Containssufficient for <n>test |  |
Temperature limit |  |
Do not reuse | |
 |
Caution | Test Device | Extraction Solution | ||
| Dropper Tip | Extraction Tube | ||||
| Positive ControlSwab | Negative ControlSwab |
![]() COVAG025-UManufacturer GenBody, Inc.3-18, Eopseong 2-Gil, Seobuk-guCheonan-si, Chungcheongnam-do, 31077, Republic of KoreaTel: +82-41-523-8993Fax: +82-41-523-8991Email: [email protected]Website: http://genbody.co.kr
COVAG025-UManufacturer GenBody, Inc.3-18, Eopseong 2-Gil, Seobuk-guCheonan-si, Chungcheongnam-do, 31077, Republic of KoreaTel: +82-41-523-8993Fax: +82-41-523-8991Email: [email protected]Website: http://genbody.co.kr
US Distributor / US Agent Kwell Laboratories, LLC3420 De Forest CircleJurupa Valley, CA 91752USATel: (949) 561-0664Email: [email protected]Website: https://kwelllabs.com/
© 2021 GenBody Inc, All rights reserved.All trademarks referenced are trademarks of GenBody Inc.
For Use Under an Emergency Use Authorization (EUA) Only.The GenBody COVID-19 Ag is an immunochromatographic rapid diagnostic test (RDT) intend e d for the qualitative detection of nucleocapsid protein antigen from SARS-CoV-2 in direct nasopharyngeal swab (NP) specimens from individuals who are suspected of COVID-19 by their healthcare provider within the first six days of symptom onset. Testing is limited to laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263 a, that meet the requirements to perform moderate, high or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., inpatient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.Results are for the identification of SARS-CoV-2 nucleocapsid antigen. Antig en is generally detectable in nasopharyngeal swab specimens during the acute phase of infection. Positive results indicate the presence of viral antigens, but clinical correlation with patient history and other diagnostic information is necessary to determine infection status. Positive results do not rule out bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of the disease. Laboratories within the United States and its territories are required to report all positive results to the appropriate public health authorities. Laboratories within the United States and its territories are required to report all results to the appropriate public health authorities.Negative results should be treated as presumptive and may be confirmed with a molecular assay, if necessary, for patient management. Negative results do not rule out SARS-CoV-2 infection and should not be used as the sole basis for treatment or patient management decisions, including infection control decisions. Negative results should be considered in the context of a patient’s recent exposures, history, and the presence of clinical signs and symptoms consistent with COVID19.The GenBody COVID-19 Ag is intended for use by medical professionals or operators trained in performing tests in point of care settings. The GenBody COVID-19 Ag is only for use under the Food and Drug Administration’s Emergency Use Authorization.The GenBody COVID-19 Ag kit contains two control swabs. Test the control swabs in the same manner as patient specimens. When the positive control is tested, reddish-purple lines appear at the C and T positions. When the negative control is tested, a reddish-purple line appears at the C position only. If external controls do not perform as expected, do not use the test results and contact Technical Support Advice Line.IMPORTANT: See Package Insert, including QC section, for complete use instructions, warnings, precautions, and limitations.Specimens should be tested as quickly as possible after specimen collection. Open the test card just prior to use, lay it flat, and perform assay as follows.
Required StatementsIn the USA, this product has not been FDA cleared or approved but has been authorizing by FDA under a EUA for use by authorizing d laboratories certified under the CLIA, 42 U.S.C.§263a, which meets requirements to perform moderate, high, or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., inpatient care settingsoperating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation. This product has been authorized only for the detection of proteins from SARS-CoV-2, not for any other viruses or pathogens. In the USA, the emergency use of this product is only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of in vitro diagnostics for the detection and/or diagnosis of the virus that causes COVID-19 under Section 564(b)(1) of the Federal Food, Drug and Cosmetic Act, 21 U.S.C. § 360bbb – 3(b)(1), unless the declaration is terminated or authorization is revoked sooner.
Contact InformationOrdering and Contact InformationUS Distributor / US Agent Kwell Laboratories, LLC3420 De Forest CircleJurupa Valley, CA 91752USATel: (949) 561-0664Email: [email protected]Website: www.kwelllabs.com
Technical Support Advice Line (US)Tel: (888) 552-5204Email: [email protected]
Part 1 – Swab Sample Collection Procedure
- Remove a nasopharyngeal swab from the pouch.
- With the patient’s head tilted backward at 70 degrees, carefully insert the swab into the nostril that presents the most secretion under visual inspection.
- Gently and slowly insert the swab through the nostril parallel to the palate (not upwards) until resistance is encountered or the distance is equivalent to that from the ear to the nostril of the patient, indicating contact with the nasopharynx.
- Leave the swab in place for several seconds to absorb secretions.
- Rotate the swab 3-5 times against the posterior nasopharynx.
- Using gentle rotation, remove the swab from the nostril; insert it into the Extraction Tube.
- All specimens should be tested as soon as they are prepared.

Refer to the CDC Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Persons for Coronavirus Disease 2019 (COVID19).
Part 2 – Test Procedure
1.
• Add the Extraction the solution to the Fill Line indicated on the Extraction Tube.
2.
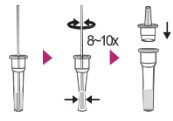
- Insert the collected specimen swab into the Extraction Solution.
- Mix by squeezing the tube and simultaneously rotating the swab 8~10 times.
- Place the Dropper Tip.
3.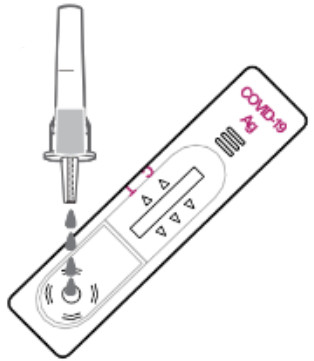
Add 4 drops of the solution to the sample well.
4.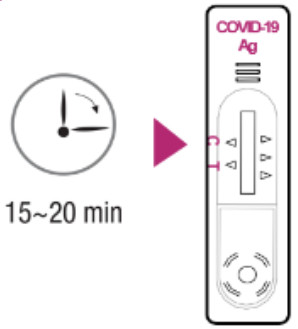
- Read the test result at 15~20 minutes.
- Do Not read the results after 20 minutes.
5.
Part 3 – Result Interpretation
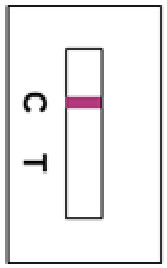
Negative ResultsOnly one reddish-purple line on the control line (C) position appears with no line on the test line position (T).
6.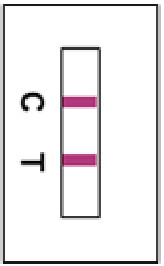
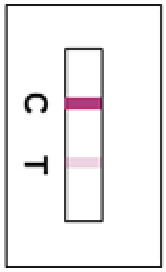
Positive ResultsTwo reddish-purple lines appear in the test window, one on the test line position (T) and the other on the control line position (C).Any faint visible reddish-purple Test line should be interpreted as positive.
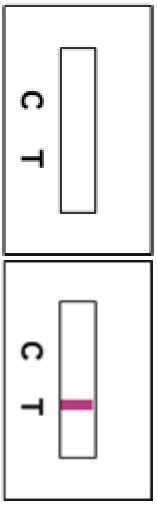
Invalid Results If a line does not appear on the control line position (C) in 15 minutes, the test result is invalid. Re-test with a new GenBody COVID-19 Ag Test Device.
![]()
3-18, Eopseong 2-gil, S eobuk -gu, Cheonan-si,Chungc heongnam-do, 31077, Republic of Korea.Tel. +82-41-523-8990+82-41-523-8993 (International)Fax. +82-41-523-8991http://www.genbody.co.kr

![]() COVAG025-U
COVAG025-U
© 2021 GenBody Inc, All rights reserved.All trademarks referenced are trademarks of GenBody Inc.
References
[xyz-ips snippet=”download-snippet”]